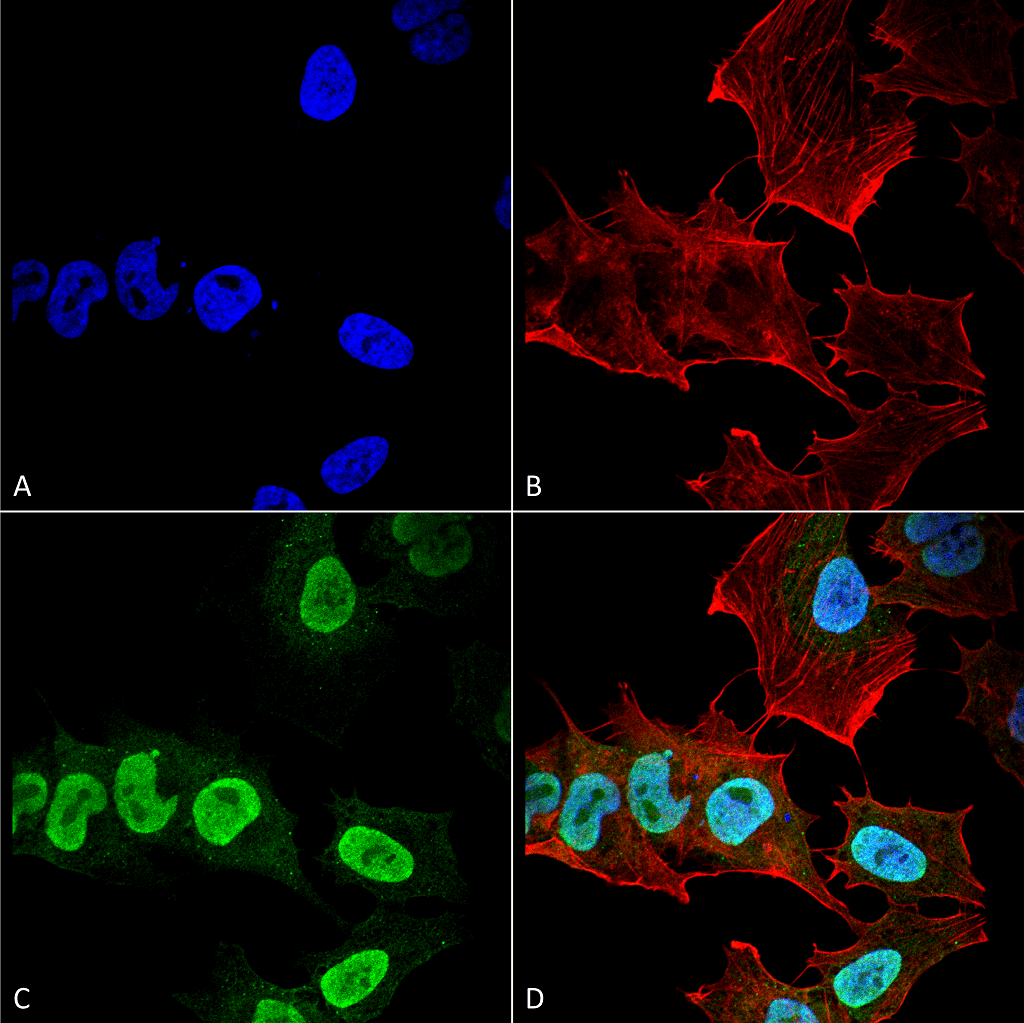
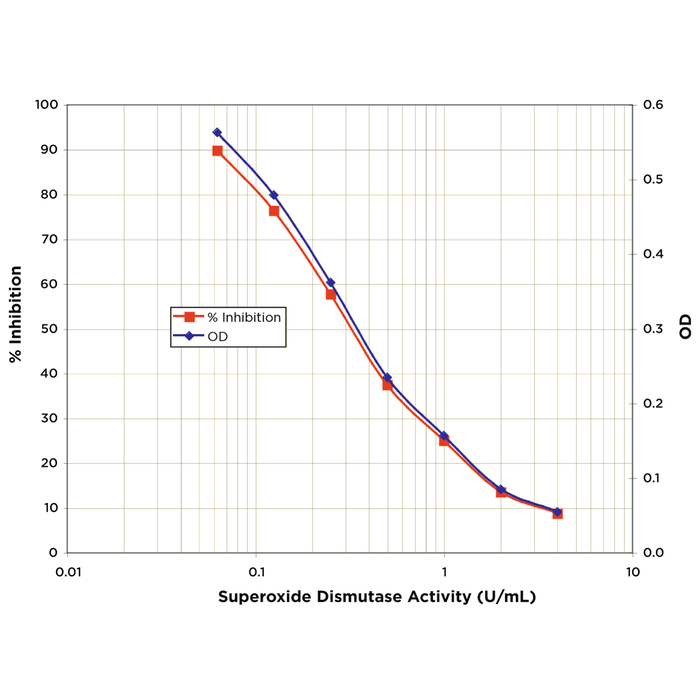
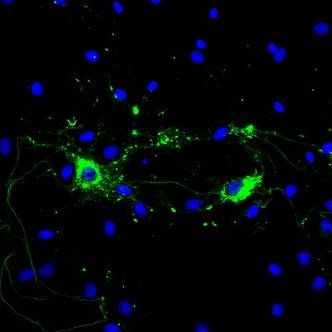
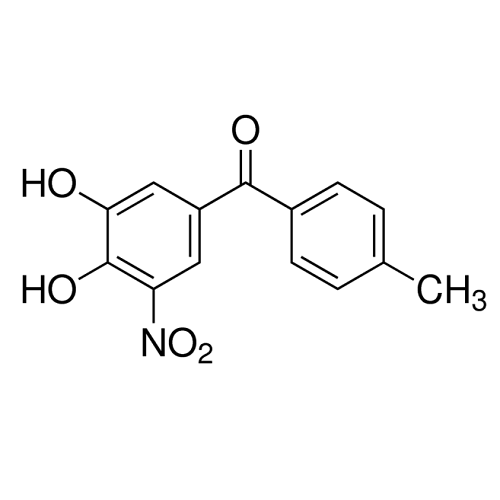
Alpha Synuclein: Inhibitors
Drug-induced inhibition of alpha synuclein aggregation and fibrilization
A common area of research in PD therapy is the development of drugs targeting fibril growth to inhibit alpha synuclein induced toxicity. Mahul-Mellier et al. (2015)., showed that Tolcapone, a fibrilization inhibitor, and the replacement of alpha synuclein with beta synuclein halts alpha synuclein toxicity in both neuroblastoma cell lines and hippocampal primary neurons. Beta synuclein lacks the NAC region (non Aβ component), therefore, does not form fibrils.
Rho-associated protein kinase (ROCK) is part of the alpha synuclein aggregation pathway and was labelled as a “novel neuroprotectant target” in PD research. Tatenhorst et al. (2016) showed that a ROCK inhibitor, Fasudil, considerably lowered α-synuclein aggregation in an H4 cell culture model and a cell-free assay. Fasudil not only inhibits ROCK activation, but directly interacts with alpha synuclein residues Y133 and Y136 in the C- terminal region. Ultimately, improved motor and cognitive functions were observed in α-SynA53T mice who received Fasudil.
Deteriorated mitochondrial function is often correlated with PD pathogenicity; Lee et al. (2002), explored the effect of mitochondrial electron-transport chain inhibitors on α-synuclein aggregation. Administration of both rotenone (complex I inhibitor) and oligomycin (ETC inhibitor) resulted in increased alpha synuclein aggregation. Therefore, alpha synuclein inclusions could be caused by impaired mitochondrial activity and reversed with the recovery of mitochondrial function.